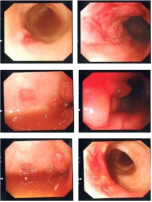
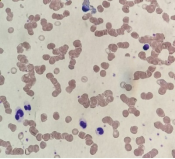
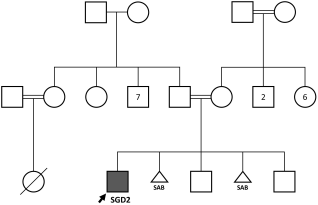
Background: Specific granulocyte deficiency type 2(SGD2) is a rare disorder of neutrophil defect caused by mutation in SMARCD2 gene characterized by neutropenia, recurrent infections, skin and mucosal ulceration. Objective: We aim to describe a symptomatic patient with a novel homozygous nonsense variant in SMARCD2 to help expand the phenotypic description of this rare disease. Methods: A retrospective chart review was conducted on our patient. Data collection included information on demographic, clinical and laboratory data. Results: We report a 4 years old boy with history of delayed separation of cord, recurrent sepsis, skin infections as well as mucocutaneous ulceration, gastrointestinal bleeding and Epstein Barr virus induced lymphoproliferation. He had variable neutropenia, bone marrow biopsy revealed hypogranular neutrophils and no blast excess. Whole genome analysis detected a novel homozygous variant in SMARCD2 gene c.208C > T p.(Gln70*). Conclusion: This case report aims to provide insight on additional clinical and molecular features of SGD2. It also sheds light on potential role of SMARCD2 gene in other immune cells.
| Published in | International Journal of Immunology (Volume 12, Issue 4) |
| DOI | 10.11648/j.iji.20241204.11 |
| Page(s) | 52-58 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2024. Published by Science Publishing Group |
Inborn Errors of Immunity, Neutropenia, SMARCD2, Specific Granulocyte Deficiency
SGD2 | Specific Granulocyte Deficiency Type 2 |
IEI | Inborn Error of Immunity |
HSCT | Hematopoietic Stem Cell Transplantation |
IVIG | Intravenous Immunoglobulin |
EBV | Epstein Barr Virus |
FTT | Failure to Thrive |
PTLD | Posttransplant Lymphoproliferative Disease |
Age | Diagnosis | Isolated organism (site) | Management |
|---|---|---|---|
2 weeks | Necrotizing fasciitis, sepsis | Pseudomonas aeruginosa (blood) | Surgical debridement, IV antibiotics and IVIG |
1.5 months | Omphalitis | - | Surgical removal and IV antibiotics |
2.5 months | Sepsis and acute gastroenteritis | Acinetobacter lwoffi (blood) Rota virus (stool) | IV antibiotics |
3 months | Sepsis, perianal ulcerative lesions and acute gastroenteritis, dehydration, electrolyte abnormalities | Rota virus (stool), Klebsiella pneumoniae (skin) | IV antibiotics |
6 months (prolonged admission) | Sepsis, chronic diarrhea, electrolyte abnormality, anemia required transfusion, pericarditis, right ileo- femoral thrombosis, Osteomyelitis of the sacroiliac, foot abscess | Enterobacter species (foot wound) | Multiple courses of antibiotics, transfusion Ibuprofen for pericarditis Enoxaparin for thrombosis |
10 months | Decompensated shock, coagulopathy electrolyte imbalance Acute gastroenteritis, upper respiratory tract infection | Pseudomonas aeruginosa (blood) Rota (stool) RSV (nasopharyngeal) | IV antibiotics, inotropic support, hydrocortisone Vitamin K and FFP |
13 & 15.5 months | Acute gastroenteritis sepsis | Negative | IV antibiotics |
16 months | Perianal abscess | Negative | IV antibiotics |
20 &22 months | Febrile neutropenia, sepsis | Negative | IV antibiotics |
24 months | Enteritis, colitis, ulcers in the ulcers seen the rectosigmoid | Negative | IV antibiotics, blood transfusion |
29 months | Colitis | Negative | IV Antibiotics and blood transfusion |
30 months | Chronic/ fungal sinusitis, mild chronic hepatitis Suspected IBD/ Chron’s, sepsis | Pseudomonas aeruginosa (sputum) Enterobacter aerogenes (blood) | IV antibiotics, IV anti-fungal, G-CSF Mesalamine |
38 months | Septic shock, EBV induced lymphoproliferative disease / HLH, MIS-C, bloody diarrhea | EBV PCR (blood), Pseudomonas aeruginosa (blood) Influenza A (nasopharyngeal aspirate) | steroids, rituximab, IVIG, blood transfusion |
Reference | Number of patients | Age of onset | Clinical features | Variant in SMARCD2 |
|---|---|---|---|---|
1 | 4 | Neonate | Delayed separation of umbilical cord, recurrent bacterial infections (pneumonia, septicemia), chronic diarrhea, anemia, thrombocytopenia, developmental delay, brittle dysplastic nails, mild distal skeletal anomalies, irregularly placed teeth, dysmorphic features | c.118+1G>A c.401+2T>C c.414_438dup |
2 | 1 | Infancy | Delayed umbilical cord separation with omphalitis, sublingual ulcer, chronic diarrhea, recurrent respiratory infections, FTT, developmental delay | c.118+1G>A |
3 | 1 | Neonate | Neonatal cholestasis, hypotonia, delayed cord separation, recurrent bacterial and fungal infections, dysmorphic features | c.511 C>T |
4 | 1 | Infancy | Delayed umbilical cord separation, recurrent infections (pneumonia and diarrhea), aphthous stomatitis, lymphadenopathy, dysmorphic features | c.93delG |
5 | 1 | Infancy | Recurrent infections (chronic rhinosinusitis, recurrent otitis media, sepsis, fungal infections, multiple superficial skin abscesses and deep cellulitis), lymphadenopathy, chronic stomatitis, vaginal, rectal ulcers, GI bleeding, post-infectious glomerulonephritis | c.217C>T c.1081del |
Our patient | 1 | Neonate | Delayed separation of umbilical cord, recurrent bacterial infections (sepsis), chronic diarrhea, recurrent ulcerative lesions including the rectum, HLH secondary to EBV infection, Lymphadenopathy, lower GI bleeding | c.208C>T |
| [1] | Witzel, M., Petersheim, D., Fan, Y., Bahrami, E., Racek, T., Rohlfs, M., Puchałka, J., Mertes, C., Gagneur, J., Ziegenhain, C., Enard, W., Stray-Pedersen, A., Arkwright, P. D., Abboud, M. R., Pazhakh, V., Lieschke, G. J., Krawitz, P. M., Dahlhoff, M., Schneider, M. R., & Wolf, E. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nature Genetics. 2017, 49(5), 742–752. |
| [2] | Schim, I., Evelien G. G. Sprenkeler, Anton T. J. Tool, Abinun, M., Grainger, A., Engelhardt, K. R., Michel van Houdt, Janssen, H., Kuijpers, T. W., & Hambleton, S. Defective neutrophil development and specific granule deficiency caused by a homozygous splice-site mutation in SMARCD2. The Journal of Allergy and Clinical Immunology. 2021, 147(6), 2381-2385. e2. |
| [3] | Kihtir, Z., Çelik, K., Tayfun Küpesiz, F., Küpesiz, O. A., Kocacik Uygun, D. F., Arayici, S., Ongun, H., Acarbulut, İ., Sağlam, C., Ceylaner, G., & Bingöl, A. Specific Granule Deficiency Due To Novel Homozygote SMARCD2 Variant. Pediatric Allergy, Immunology, and Pulmonology. 2022, 35(1), 43–46. |
| [4] | Esra Yücel, İbrahim Serhat Karakuş, Krolo, A., Ayça Kıykım, Raúl Jimenez Heredia, Tamay, Z., Funda Çipe, Elif Karakoç-Aydıner, Ahmet Özen, Karaman, S., Kaan Boztuğ, & Safa Barış. Novel Frameshift Autosomal Recessive Loss-of-Function Mutation in SMARCD2 Encoding a Chromatin Remodeling Factor Mediates Granulopoiesis. Journal of Clinical Immunology. 2020, 41(1), 59–65. |
| [5] | Ibrahim, A., Anjali Sharathkumar, McLaughlin, H., Claassen, D. A., & Sharathkumar Bhagavathi. Congenital Neutropenia with Specific Granulocyte Deficiency Caused by Novel Double Heterozygous SMARCD2 Mutations. Hematology Reviews. 2022, 14(3), 270–275. |
| [6] | Ali, A., Tabouni, M., Praseetha Kizhakkedath, Baydoun, I., Allam, M., John, A., Faiza Busafared, Alnuaimi, A., Fatma Al-Jasmi, & Hiba Alblooshi. Spectrum of genetic variants in bilateral sensorineural hearing loss. Frontiers in Genetics. 2024, 15. |
| [7] | Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S. T., Abeysinghe, S., Krawczak, M., & Cooper, D. N. Human Gene Mutation Database (HGMD®): 2003 update. Human Mutation. 2003, 21(6), 577–581. |
| [8] | Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P., Gauthier, L. D., Brand, H., Solomonson, M., Watts, N. A., Rhodes, D., Singer-Berk, M., England, E. M., Seaby, E. G., Kosmicki, J. A., & Walters, R. K. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020, 581(7809), 434–443. |
| [9] | Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2015, 17(5), 405–424. |
| [10] | GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, N. Y.). 2015, 348(6235), 648–660. |
| [11] | Priam, P., Krasteva, V., Rousseau, P., D’Angelo, G., Gaboury, L., Sauvageau, G., & Lessard, J. A. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPɛ dependent mechanism. Nature Genetics. 2017, 49(5), 753–764. |
| [12] | Ruo Ran Wang, Pan, R., Zhang, W., Fu, J., Lin, J. D., & Zhuo Xian Meng. The SWI/SNF chromatin-remodeling factors BAF60a, b, and c in nutrient signaling and metabolic control. Protein & Cell. 2017, 9(2), 207–215. |
| [13] | Cohen, J. I., & Meyts, I. Editorial: EBV Infection and Human Primary Immune Deficiencies. Frontiers Immunology. 2020, 11. |
| [14] | Ru, Y., Chen, J., & Wu, D. Epstein‐Barr virus post‐transplant lymphoproliferative disease (PTLD) after hematopoietic stem cell transplantation. European Journal of Haematology. 2018, 101(3), 283–290. |
| [15] | Sharma, S., Rakesh Kumar Pilania, Gummadi Anjani, Sudhakar, M., Arora, K., Tyagi, R., Dhaliwal, M., Pandiarajan Vignesh, Rawat, A., & Singh, S. Lymphoproliferation in Inborn Errors of Immunity: The Eye Does Not See What the Mind Does Not Know. Frontiers in Immunology. 2022, 13. |
| [16] | Colucci, M., Carsetti, R., Serafinelli, J., Rocca, S., Massella, L., Gargiulo, A., Lo Russo, A., Capponi, C., Cotugno, N., Porzio, O., Onetti Muda, A., Palma, P., Emma, F., & Vivarelli, M. Prolonged Impairment of Immunological Memory After Anti-CD20 Treatment in Pediatric Idiopathic Nephrotic Syndrome. Frontiers in Immunology. 2019, 10. |
| [17] | Makatsori, M., Kiani-Alikhan, S., Manson, A. L., Verma, N., Leandro, M., Gurugama, N. P., Longhurst, H. J., Grigoriadou, S., Buckland, M., Kanfer, E., Hanson, S., Ibrahim, M. A. A., Grimbacher, B., Chee, R., & Seneviratne, S. L. Hypogammaglobulinaemia after rituximab treatment--incidence and outcomes. QJM. 2014, 107(10), 821–828. |
| [18] | Fournier, B. M., & Parkos, C. A. The role of neutrophils during intestinal inflammation. Mucosal Immunology. 2012, 5(4), 354–366. |
| [19] | Yamamoto-Furusho, J. K. (2006). Crohn’s disease: Innate immunodeficiency? World Journal of Gastroenterology, 12(42), 6751. |
APA Style
Kuwaiti, A. A. A., Awadhi, H. A., Kizhakkedath, P., Baydoun, I., Tabouni, M., et al. (2024). A Novel Nonsense Homozygous Mutation in SMARCD2 Gene Causing Specific Granulocyte Deficiency Type 2: Case Report. International Journal of Immunology, 12(4), 52-58. https://doi.org/10.11648/j.iji.20241204.11
ACS Style
Kuwaiti, A. A. A.; Awadhi, H. A.; Kizhakkedath, P.; Baydoun, I.; Tabouni, M., et al. A Novel Nonsense Homozygous Mutation in SMARCD2 Gene Causing Specific Granulocyte Deficiency Type 2: Case Report. Int. J. Immunol. 2024, 12(4), 52-58. doi: 10.11648/j.iji.20241204.11
@article{10.11648/j.iji.20241204.11,
author = {Amna Ali Al Kuwaiti and Haifa Al Awadhi and Praseetha Kizhakkedath and Ibrahim Baydoun and Mohammed Tabouni and Hiba Alblooshi and Fatma Al-Jasmi and Aisha Al Shamsi and Hiba Mohammed Shendi},
title = {A Novel Nonsense Homozygous Mutation in SMARCD2 Gene Causing Specific Granulocyte Deficiency Type 2: Case Report
},
journal = {International Journal of Immunology},
volume = {12},
number = {4},
pages = {52-58},
doi = {10.11648/j.iji.20241204.11},
url = {https://doi.org/10.11648/j.iji.20241204.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.iji.20241204.11},
abstract = {Background: Specific granulocyte deficiency type 2(SGD2) is a rare disorder of neutrophil defect caused by mutation in SMARCD2 gene characterized by neutropenia, recurrent infections, skin and mucosal ulceration. Objective: We aim to describe a symptomatic patient with a novel homozygous nonsense variant in SMARCD2 to help expand the phenotypic description of this rare disease. Methods: A retrospective chart review was conducted on our patient. Data collection included information on demographic, clinical and laboratory data. Results: We report a 4 years old boy with history of delayed separation of cord, recurrent sepsis, skin infections as well as mucocutaneous ulceration, gastrointestinal bleeding and Epstein Barr virus induced lymphoproliferation. He had variable neutropenia, bone marrow biopsy revealed hypogranular neutrophils and no blast excess. Whole genome analysis detected a novel homozygous variant in SMARCD2 gene c.208C > T p.(Gln70*). Conclusion: This case report aims to provide insight on additional clinical and molecular features of SGD2. It also sheds light on potential role of SMARCD2 gene in other immune cells.
},
year = {2024}
}
TY - JOUR T1 - A Novel Nonsense Homozygous Mutation in SMARCD2 Gene Causing Specific Granulocyte Deficiency Type 2: Case Report AU - Amna Ali Al Kuwaiti AU - Haifa Al Awadhi AU - Praseetha Kizhakkedath AU - Ibrahim Baydoun AU - Mohammed Tabouni AU - Hiba Alblooshi AU - Fatma Al-Jasmi AU - Aisha Al Shamsi AU - Hiba Mohammed Shendi Y1 - 2024/12/31 PY - 2024 N1 - https://doi.org/10.11648/j.iji.20241204.11 DO - 10.11648/j.iji.20241204.11 T2 - International Journal of Immunology JF - International Journal of Immunology JO - International Journal of Immunology SP - 52 EP - 58 PB - Science Publishing Group SN - 2329-1753 UR - https://doi.org/10.11648/j.iji.20241204.11 AB - Background: Specific granulocyte deficiency type 2(SGD2) is a rare disorder of neutrophil defect caused by mutation in SMARCD2 gene characterized by neutropenia, recurrent infections, skin and mucosal ulceration. Objective: We aim to describe a symptomatic patient with a novel homozygous nonsense variant in SMARCD2 to help expand the phenotypic description of this rare disease. Methods: A retrospective chart review was conducted on our patient. Data collection included information on demographic, clinical and laboratory data. Results: We report a 4 years old boy with history of delayed separation of cord, recurrent sepsis, skin infections as well as mucocutaneous ulceration, gastrointestinal bleeding and Epstein Barr virus induced lymphoproliferation. He had variable neutropenia, bone marrow biopsy revealed hypogranular neutrophils and no blast excess. Whole genome analysis detected a novel homozygous variant in SMARCD2 gene c.208C > T p.(Gln70*). Conclusion: This case report aims to provide insight on additional clinical and molecular features of SGD2. It also sheds light on potential role of SMARCD2 gene in other immune cells. VL - 12 IS - 4 ER -
Division of Pediatric Allergy / Immunology, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates
Division of Pediatric Gastroenterology, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates
Department of Genetics and Genomics, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates; Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates
Division of Genetics, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates
Division of Pediatric Allergy / Immunology, Department of Pediatrics, Tawam Hospital, Al-Ain, United Arab Emirates